Un attuale dilemma diagnostico
ABSTRACT
Nonostante L’Alzheimer sia la forma più comune di demenza, nonché venga riconosciuta come una delle patologie a più grave impatto sociale, la sua causa e la progressione non sono ancora chiaramente definiti. Un’ ipotesi comunemente condivisa individua nella formazione di placche di amiloide e nell’agglomerazione di ammassi neurofibrillari la probabile origine della malattia. In contrasto, è stata recentemente vagliata l’ipotesi secondo cui la malattia di Alzheimer sia da considerare una patologia vascolare.
« Ogni qualvolta una teoria ti sembra essere l’unica possibile,
prendilo come un segno che non hai capito né la teoria
né il problema che si intendeva risolvere. »
(Karl Popper, Conoscenza oggettiva: un punto di vista evoluzionistico.)
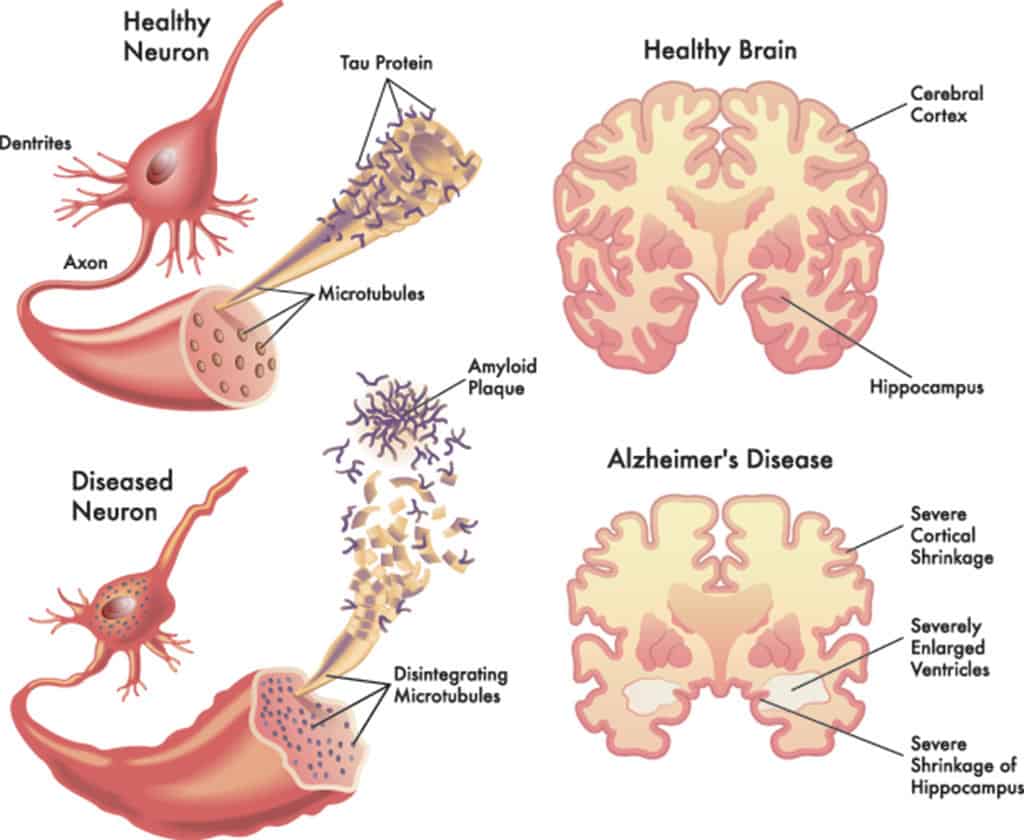
 La malattia di Alzheimer (AD) è una malattia degenerativa progressiva caratterizzata dalla perdita di funzione e morte delle cellule neuronali in diverse aree del cervello. E’ la forma più comune di demenza, progressivamente invalidante e con esordio prevalentemente in età presenile (>65 anni) anche se può svilupparsi in epoca precedente. Nel 2006 il numero dei malati di Alzheimer nel mondo è stato stimato in 26,6 milioni ed in proiezione si ipotizza che ne sarà affetta 1 persona su 85 nel 2050 (Brookmeyer et al. 2007). La caratteristica macroscopica più evidente del cervello di un soggetto affetto da malattia di Alzheimer è la marcata atrofia che determina un’aumentata ampiezza dei solchi cerebrali e l’incremento del volume ventricolare.
La malattia di Alzheimer (AD) è una malattia degenerativa progressiva caratterizzata dalla perdita di funzione e morte delle cellule neuronali in diverse aree del cervello. E’ la forma più comune di demenza, progressivamente invalidante e con esordio prevalentemente in età presenile (>65 anni) anche se può svilupparsi in epoca precedente. Nel 2006 il numero dei malati di Alzheimer nel mondo è stato stimato in 26,6 milioni ed in proiezione si ipotizza che ne sarà affetta 1 persona su 85 nel 2050 (Brookmeyer et al. 2007). La caratteristica macroscopica più evidente del cervello di un soggetto affetto da malattia di Alzheimer è la marcata atrofia che determina un’aumentata ampiezza dei solchi cerebrali e l’incremento del volume ventricolare.
Questa atrofia appare diffusa interessando, oltre al lobo temporale, le aree associative corticali, l’ippocampo ed il giro para-ippocampale, con un relativo risparmio delle aree posteriori degli emisferi, del cervelletto e del tronco cerebrale. L’atrofia è legata principalmente alla degenerazione neuronale, che comporta la riduzione del numero di spine dendritiche e di giunzioni sinaptiche, fino ad una vera e propria scomparsa della cellula nervosa, fenomeno questo che si determinerebbe con un meccanismo apoptotico.
Una delle maggiori sfide in campo scientifico è da sempre stata quella di chiarire cosa causi la malattia di Alzheimer. Conoscerne il substrato eziopatogenetico condurrebbe ad indubbi benefici nella pratica clinica e nello sviluppo di un efficace trattamento.
Tra le ipotesi più accreditate per decenni vi è l’ “ipotesi amiloide”, proposta per la prima volta a metà degli anni ottanta da Masters e colleghi (Master set al. 1985) i quali evidenziarono che le placche senili presenti nel tessuto cerebrale di soggetti con AD fossero costituite principalmente da un peptide denominato β-amiloide (A β), un derivato della proteina APP (Amyloid Precursor Protein). Secondo tale ipotesi l’evento patogenetico chiave responsabile della degenerazione dei neuroni e delle modificazioni morfologiche, funzionali e cognitive tipiche dell’ Alzheimer è, di fatto, l’eccessiva formazione o accumulo di peptidi amiloidogenetici (Hardy, Selkoe, 2002).
Sono sostanzialmente due i meccanismi attraverso i quali la β-amiloide produce danni neuronali e funzionali:
– Un meccanismo diretto, nel quale Aβ interagisce con componenti della membrana cellulare e danneggia direttamente i neuroni e/o aumenta la suscettibilità dei neuroni ad una varietà di fattori di danno, come l’eccitotossicità, l’ipoglicemia o il danno perossidativo (Koh et al. 1990).
– Un meccanismo indiretto, nel quale Aβ danneggia i neuroni indirettamente tramite l’attivazione della microglia e degli astrociti a produrre mediatori tossici ed infiammatori, come ad esempio l’ossido nitrico, le citochine e gli intermedi reattivi dell’ossigeno che causano la morte dei neuroni per apoptosi o per necrosi (Meda et al. 1995; Della Bianca et al. 1999)
Negli ultimi anni si sono accumulate prove con modelli animali (si vedano ad esempio: Hsiao et al. 1996; Koistinaho et al 2001; Irizarry et al. 1997) che mettono in dubbio la validità dell’ipotesi della Aβ nella sua enunciazione primitiva, cioè che i deficit cognitivi dell’Alzheimer siano dovuti all’effetto citotossico che si verifica a livello delle placche da parte della Aβ in forma fibrillare. La critica principale all’ipotesi amiloide è che i deficit funzionali, cioè perdita della memoria, deficit cognitivi oppure deficit di apprendimento, non correlano né temporalmente né quantitativamente con la formazione delle placche e quindi con l’accumulo di Aβ in forma fibrillare (Querfurth, La Ferla, 2010). Inoltre il deposito di Aβ nel cervello non è stato dimostrato essere neurotossico in vivo (de la Torre, 2004); sembra altresì non essere direttamente correlato con la gravità della demenza (Terry et al. 1991) ed in molti pazienti non affetti da demenza sono state riscontrate le medesime placche senili dell’AD (Arrigada et al. 1992). Ulteriore limite dell’ipotesi amiloide è infine rappresentato dalla mancanza di effettivi trattamenti farmacologici disponibili che supportino tale substrato teorico (de la Torre 2002).
Alla luce di queste criticità riscontrate, sono state negli anni formulate versioni alternative dell’ipotesi amiloide secondo le quali il ruolo chiave della Aβ nella patogenesi della malattia sarebbe mantenuto, ma la forma patogena non sarebbe solo quella fibrillare, ma anche altre quali piccoli oligomeri solubili e protofibrille, cioè forme precedenti la loro aggregazione in forma fibrillare (Koh et al. 1990).
Un nuovo recente studio (Jonsson et al. 2012) rilancia invece l’ipotesi amiloide grazie alla scoperta di una mutazione in un gene chiave della proteina amiloide, in grado di proteggere dallo sviluppo della malattia: le persone anziane portatrici di una certa variante del gene APP, che codifica per la proteina precursore della beta-amiloide, hanno una probabilità decisamente inferiore di ammalarsi di Alzheimer o di andare incontro al declino cognitivo legato all’età.
E’ evidente che, nonostante l’ipotesi amiloide sia stato uno dei modelli di riferimento più influenti delle ricerche sulla patogenesi della malattia di Alzheimer, il ruolo della Aβ nello sviluppo della sintomatologia clinica rimanga ancora sotto la lente di ingrandimento (per un’analisi critica: Armstrong R. 2014; Morris et al. 2014).
Continua a destare un sempre più crescente interesse un secondo filone di ricerca che fa riferimento ad un possibile substrato vascolare in merito alla patogenesi della malattia di Alzheimer.
Studi epidemiologici, trial clinici e studi basati su modelli animali condividono ormai l’idea generale che malattie cerebrovascolari conducono, in differenti modalità, al declino cognitivo (Gorelik et al. 2011). Diversi fattori di rischio per patologie vascolari, quali ipertensione, diabete mellito, aterosclerosi, ipercolesterolemia, obesità, sindromi metaboliche (si veda Kalaria et al. 2012, per una visione d’insieme), sono stati altresì associati specificamente anche alla demenza di Alzheimer. Senza dimenticare che già i referti autoptici del primo vero documentato paziente con diagnosi di AD evidenziarono la presenza di una patologia micro vascolare (Alzheimer, 1907). E’ inoltre stato proposto che una riduzione del fluido cerebrospinale (CSF, cerebrospinal fluid) possa essere considerata un potenziale biomarker per la malattia di Alzheimer (Mazza et al. 2011).
“L’ipotesi vascolare” si basa sull’idea che la presenza di una patologia vascolare che coinvolge rigidità arteriosa, arteriosclerosi, degenerazione dell’endotelio e disfunzioni della barriera emato-encefalica, conduca conseguentemente ad una ipoperfusione cerebrale cronica (Kalaria et al. 2012).Proprio tale condizione sarebbe connessa alla marcata atrofia cerebrale ed all’accumulo della proteina beta-amiloide (Velliquette et al. 2005), tipici della malattia di Alzheimer, ed è stata rilevata anche in pazienti in uno stadio pre-clinico (Mild Cognitive Impairment – Deterioramento Cognitivo Lieve) così come in soggetti sani a rischio di sviluppare AD a causa di fattori familiari e/o genetici (Austin et al. 2011).
Recenti studi interessati ad investigare il CSF in soggetti con Alzheimer hanno di fatto evidenziato che condizioni quali l’ipoperfusione siano associate all’AD molto più frequentemente di quanto si pensasse in passato (Chen et al. 2011).
Investigare il grado di perfusione in soggetti con Alzheimer è tuttavia un compito non semplice, dal momento che la riduzione del CSF è solo una delle molteplici componenti associate all’ AD. La contemporanea presenza di ipoperfusione, placche arteriose, grovigli neuro-fibrillari, depositi di amiloide, atrofia e stenosi complica non poco la distinzione tra cause ed effetti (Austin et. al 2011).
Esiste un ampio range di tecniche di neuroimaging finalizzate allo studio della malattia di Alzheimer. Recentemente sta acquisendo sempre più consenso una tecnica di risonanza magnetica non invasiva denominata arterial spin labeling (ASL), che consiste nel marcare magneticamente l’afflusso prossimale di sangue affluente ad una determinata area di tessuto: in questo caso, il segnale rilevato nel tessuto è proporzionale al flusso sanguigno, fornendo in tal modo una misura di perfusione. Uno dei vantaggi di questa tecnica è che è in grado di fornire una misura quantitativa di fluido cerebrospinale piuttosto che una misura relativa, come nel caso del segnale BOLD (Blood Oxygen Level Dependent) nella risonanza magnetica funzionale (Liu, Brown, 2007).
La valutazione in vivo del CSF mediante la arterial spin labeling può ritenersi pertanto un promettente strumento utile per la diagnosi precoce e per la descrizione della progressione della malattia di Alzheimer (Wierenga et al. 2014). Diversi studi nei quali è stata impiegata la ASL hanno inoltre evidenziato che l’Alzheimer sia associato ad ipoperfusione sia globale che specifica per alcune regioni ed hanno altresì rilevato una considerevole sovrapposizione tra le regioni cerebrali associate alla malattia e quelle associate allo stato di rischio. (Alsop et al. 2010)
In conclusione, l’idea che la genesi della malattia di Alzheimer sia connessa ad una patologia vascolare, piuttosto che riconducibile alla sola ipotesi amiloide appare sempre più condivisa dalla comunità scientifica, suffragata da molteplici studi epidemiologici e su animali. Terapie farmacologiche impiegate in soggetti affetti da patologie vascolari sembrerebbero inoltre avere benefici anche in soggetti con AD (Kalaria et al. 2012).
Tuttavia esistono ancora diversi aspetti che necessitano di essere ulteriormente validati riguardanti la natura effettiva di causa-effetto tra le due patologie concomitanti.
BIBLIOGRAFIA
Alsop DC., Dai W., Grossman M., Detre JA. Arterial spin labeling blood flow MRI: its role in the early characterization of Alzheimer’s disease. J Alzheimers Dis 2010; 20:871-880.
Alzheimer A. Uber eine eigenartig Erkrankung er Hirnrinde. Allg Z Psychiat Psych-Gerichtl Med 1907; 64:146-8.
Armstrong R.A. A critical analysis of the ‘amyloid cascade hypothesis’. Folia Neuropathol. 2014; 52 (3): 211-225.
Arriagada PV., Growdon JH., Hedley-White ET., Hyman B. Neurofibrillar tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992; 42: 631–39.
Austin B.P., Nair V.A., Meier T.B., Xu G., Rowley H.A., Carlsson C.M., Johnson S.C., Prabhakaran V. Effects of Hypoperfusion in Alzheimer’s Disease. Journal of Alzheimer’s Disease . 2011; 26:123–133.
Brookmeyer R., Johnson E., Ziegler-Graham K., Arrighi H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s & Dementia. 2007; 3:186–191.
Chen W., Song X., Beyea S., D’Arcy R., Zhang Y., Rockwood K. Advances in perfusion magnetic resonance imaging in Alzheimer’s disease. Alzheimers Dement 2011; 7:185-196.
de la Torre J. C. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004; 3:184-190
de la Torre JC. Alzheimer’s disease as a vascular disorder: nosological evidence. Stroke 2002; 33: 1152–62.
Della Bianca V., Dusi S., Bianchini E., Dal Pra I., Rossi F. β-amyloid activates O2-forming NADPH oxidase in microglia, monocytes and neutrphils. A possibile inflammatory mechanism of neuronal damage in Alzheimer’s disease. J Biol Chem. 1999; 274: 15493-15499.
Gorelik PB., Scuteri A., Black SE., Decarli C., Greenberg SM., Iadecola C., Launer LJ., Launer S., Lopez OL., Nyenhuis D., Petersen RC., Schneider JA., Tzourio C., Arnett DK., Bennett DA., Chui HC., Higashida RT., Lindquist R., Nilsson PM., Roman GC., Sellke FW., Seshadri S., American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011; 42(9):2672-713.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002; 297:353-356.
Hsiao K., Champman P., Nilsen S., Eckman C., Harigawa Y., Younkin S., Yang F., Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996; 274:99-102.
Irizarry MC., McNamara M., Fedorchak K., Hsiao K., Hyman BT. PPSw transgenic mice develop age-related A- beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997; 56: 965–73.
Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R.R., Huttenloche J., Bjornsdottir G., Andreassen O.A., Johnsson E.G., Palotie A., Behrens T.W., Magnusson O.T., Kong A., Thorsteinsdottir U., Watts R.J., Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012; 488.
Kalaria R.N., Akinyemi R., Ihara M. Does vascular pathology contribute to Alzheimer changes? Journal of the Neurological Sciences 2012; 322:141–147.
Koh JY., Yang LL., Cotman CW. β -amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res. 1990; 533:315-320.
Koistinaho M., Ort M., Cimadevilla JM., Vondrous R., Cordell B., Koistinaho J., Bures J., Higgins LS. Specific spatial learning deficits become severe with age in beta -amyloid precursor protein transgenic mice that harbor diffuse beta -amyloid deposits but do not form plaques. Proc Natl Acad Sci USA. 2001; 98:14675–80.
Liu TT., Brown GG. Measurement of cerebral perfusion with arterial spin labeling: Part 1. Methods J Int Neuropsychol Soc 2007; 13:517-525.
Masters CL., Simms G., Weinman NA., Multhaup G., McDonald BL., Beyreuther K. Amyloid plaque core protein in Alzheimer’s disease and Down Syndrome. Proc. Natc. Acad. Sci. USA. 1985; 82:4245-4249.
Mazza M., Marano G., Traversi G., Bria P., Mazza S. Primary cerebral blood flow deficiency and Alzheimer’s disease: shadows and lights. J Alzheimers Dis 2011; 23:375-389.
Meda L., Cassatella MA., Szendrei GI., Otvo L Jr., Baron P., Villalba M., Ferari D., Rossi F. Activation of microglia cells by β-amyloid protein and interferon-γ. Nature. 1995; 374:647-650.
Morris G.P., Clark I.A., Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s Disease.
Querfurth HW., La Ferla FM. Alzheimer’s disease. N Engl J Med. 2010; 362:329-344.
Terry RD., Masliah E., Salmon DP:, Butters N., De Teresa R., Hill R., Hansen LA., Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991; 4: 572–80.
Velliquette RA., O’Connor T., Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J Neurosci 2005; 25(47):10,874-83.
Wierenga C.E., Hays C.C:, Zlatar Z.Z. Cerebral blood flow measured by Arterial Spin Labeling MRI as a preclinical marker of Alzheimer’s Disease. Journal of Alzheimer’s Disease 2014; 42:411-419.
Zhao Y., Gong CX. From Chronic Cerebral Hypoperfusion to Alzheimer-like brain pathology and neurodegeneration. Cell Mol Neurobiol. 2014. [Epub ahead of print]
L’Alzheimer dovrebbe essere originato dall’incapacità del neurone di riuscire a mantenere le precise proporzioni di alcune proteine al suo interno in alcuni momenti di massima attività,perdendo così la propria omeostasi.
Questo è dovuto probabilmente a uno squilibrio nel suo funzionamento,dove lunghi periodi di bassa attività si alternano con dei momenti di attività massima,rendendo in questo modo difficile al neurone programmare le dimensioni e il lavoro delle strutture interne che gestiscono la produzione e lo stoccaggio di alcune proteine,da impiegare a tempo debito nel loro turnover ,e permettere così il corretto funzionamento della cellula.
I neuroni che si trovano in difficoltà in alcuni momenti della loro attività,dovrebbero essere quelli che hanno partecipato all’assemblaggio e alla maturazione del cervello,ma che in seguito sono diventati praticamente inutili.Questi neuroni dovrebbero essere inseriti a macchia di leopardo in un circuito che,parte dal setto, continua nell’ippocampo, nel subiculum,la corteccia entonrinale,le aree limbiche e va a chiudersi nelle aree 9, 7 ,e 22 della corteccia cerebrale.
I neuroni incriminati possono avere il loro punto debole negli assoni collaterali che hanno partecipato alla costruzione della corteccia cerebrale embrionale .
L’effetto dello squilibrio omeostatico interno al neurone provoca una modifica nella proteina TAU con una formazione e aggregazione di grovigli ,e conseguente disgregazione del microtubulo.
Questi grovigli di TAU quando assumono dimensioni significative,sono in grado di esercitare una forza attrattiva,che possiamo chiamare “elettrostatica”,sulla beta-amiloide che si trova nelle vicinanze della membrana plasmatica,favorendo l’aggregazione e l’accumulo continuo di quest’ultima fino a raggiungere una massa proporzionata al groviglio di TAU nel citoplasma,questa massa destabilizza ulteriormente l’assone,il quale come conseguenza finale porterà alla morte il neurone.
Le placche di beta-amiloide che vengono prodotte,se superano una densità critica all’interno dell’ippocampo e del subiculum,diventano tossiche per i neuroni nelle vicinanze,e capaci di destabilizzare le frequenze cerebrali,innescando in questo modo un “contagio”che coinvolge progressivamente altre aree corticali.
“L‘ipotesi vascolare”dovrebbe essere valida ,in relazione alla genesi dell’Alzheimer,solo per le due aree cerebrali dette sopra ,cioè l’ippocampo e il subiculum.
Per provare a contrastare questo possibile “contagio”,bisognerebbe intervenire prima che questo si sviluppasse,cercando di congelare lo sviluppo delle placche di beta-amiloide,e con questo la regolarità del funzionamento di tutto il neurone.
Dato che modificare dall’interno ogni neurone a rischio “contagio” è praticamente impossibile,si può solo cercare di intervenire sulla beta-amiloide,come i numerosi progetti di ricerca in cantiere stanno a dimostrare.
L’ideale sarebbe; progettare ed assemblare una proteina che cerchi di annullare la carica “elettrostatica”che esercita la placca di beta-amiloide sul citoplasma del neurone con cui è in contatto,per fare questo la proteina dovrebbe essere in grado di; agganciarsi alla placca di beta-amiloide in formazione e alla membrana plasmatica,e produrre un ammasso sufficiente a contrastare l’effetto dell’amiloide,annullando così la tossicità di quest’ultima,e permettendo al neurone di funzionare regolarmente.
In apparenza sembra che,al momento,i biologi non abbiano ancora le idee chiare su come si possa assemblare una proteina complessa;se questo fosse vero, parlare addirittura di progettare una proteina ad hoc per una determinata funzione sembra pura fantascienza.
Dunque non resta che sperare nei rapidi progressi della biologia,e che si possa un giorno non troppo lontano,trovare un modo per contrastare la più devastante delle malattie neurodegenerative.
Fiorenzo Masotti
P.S.
Avendo studiato in modo intuitivo un po di chimica e di biologia in internet,e lo stesso discorso può valere per le neuroscienze,mi scuso se ho detto qualche castroneria.
Rileggendo il commento precedente,mi sono reso conto di essere stato troppo superficiale e frettoloso nella conclusione.
Nel commento avevo dato per scontata l’ipotesi che degradare la beta-amiloide fosse praticamente impossibile o controproducente.
La beta-amiloide dovrebbe avere delle caratteristiche analogiche energetiche simili alla membrana plasmatica.
Ogni tentativo di una sua degradazione,va a impattare con un possibile danneggiamento della membrana cellulare,o di un suo alterato funzionamento.
Fiorenzo Masotti
Gli stimoli catalitici con la marcata atrofia e l’aumento dei solchi cerebrali hanno poco a che fare ma l’aumento del volume ventricolare che possono essere oggetti di degenerazione neuronale con la conseguenza di non rigenerazione cosa che se attivi i sensori cataliti le giunzioni sinaptiche fino a d un assorbimento atrofico della cellula , fungendo da precursore della cellula neuronale BCL2 con la conseguente conoscenza dell’ezio-
logia dell’ipotesi amiloidea potrebbe capovolgere il degrado.
Una sequenza parziale di un peptide 4KDaB e la troppa esposizione all’acido glutammico crea un affanno portando ad una sincope cerebrale ma la sincope non necessariamente risulta recidivà a differenza della malattia di Alzheimer.
L’idea che macibig sia un provocatore volutamente confusionario,e forse anche di mia conoscenza, dotato di uno stile letterario inconfondibile e di una fantasia illimitata,mi stimola ad approfondire l’argomento alzheimer; a proposito di fantasia illimitata,faccio notare che tra tutte le categorie di artisti quella dei,poeti scrittori,ha la percentuale maggiore di schizofrenici .
L’alzheimer forse si potrebbe curare con gli stessi sistemi con cui si cura l’epilessia,cioè con farmaci e l’ablazione di mirate aree corticali,anche se in questo caso si dovrebbe intervenire più selettivamente sopra aree estese di corteccia cerebrale.
Se l’invecchiamento procura un deficit non più rimediabile in un circuito coinvolto nel funzionamento dell’ippocampo,diciamo un abbassamento di attività sotto la soglia necessaria per svolgere correttamente il suo lavoro,invece che tentare, di ripristinarlo a oltranza o altre soluzioni,mettendo in conto dei tempi molto lunghi,si potrebbe cercare una “soluzione più rapida”,sempre prima che il danno diventi irreparabile,cioè cercare di abbassare l’attività dei circuiti con cui lavora quello “spompato“ entrato in crisi,in modo da garantire un’attività ridotta ma ancora possibile di tutto l‘ippocampo.
Forse questo si potrebbe fare ,in qualche modo,sfoltendo i fasci di neuroni in entrata e uscita dall’ippocampo,fisicamente o con medicinali.
Fiorenzo Masotti